
Kevin L. Ong, Ph.D., P.E.1; Joshua White, Ph.D.2
1 Principal Engineer, Biomedical Engineering practice, Exponent, Inc., Philadelphia, PA
2 Senior Associate, Biomedical Engineering practice, Exponent, Inc., Philadelphia, PA
The United States Food and Drug Administration (FDA) conducts inspections of manufacturers in the life sciences industry for various reasons. Potential violations of the FDA’s requirements can lead to Inspectional Observations (Form 483s) or Warning Letters. Inadequate responsiveness to FDA Warning Letters may lead to severe repercussions, such as product seizures, delay in regulatory approvals/clearances, and even civil penalties.
Quality assurance in the drug and medical device industries
In recent years, healthcare needs have grown exponentially, including the use of drugs and medical devices to address health conditions for the growing, aging population in the U.S. There has also been an increasing need for treatment options to address similarly increasing rates of chronic conditions, such as obesity and diabetes, which affect populations of all ages. These factors, among others, have spurred growth in the research and development of pharmaceutics, diagnostics, and medical technologies. In turn, there has been a corresponding increase in governmental regulation and oversight of these technologies.
The FDA provides regulatory oversight to the healthcare industry in terms of safety and efficacy and compliance with quality systems. The requirements for quality systems are guided by the Quality Systems Regulations (QSR or 21 C.F.R. § 820), which became effective in 1997 and replaced the previous 1978 Good Manufacturing Practices (GMP) for medical devices. The updated regulation added design controls (described later) after the FDA found that 44% of quality issues leading to voluntary recall actions were attributable to errors or deficiencies in product design that may have been prevented by adequate design controls. In essence, the QSR provides a framework for companies to achieve quality requirements that relate to designing, manufacturing, packaging, labeling, storing, installing, and servicing of pharmaceutical drugs and medical devices.
FDA inspections
The FDA assesses a company’s compliance with quality regulations through inspections and audits. The FDA inspects manufacturers of finished medical devices and drugs as well as vendors/suppliers of components, parts, or accessories of devices. The inspections include domestic and foreign sites that produce FDA-regulated products or components that are imported into and/or sold within the U.S. as well as domestic sites that import and distribute foreign-manufactured goods within the U.S. Routine and periodic inspections are conducted, but they can also be performed at any time for reasons such as:
- To investigate problems brought to the FDA’s attention regarding product safety or commercial fraud (i.e., for cause);
- To verify appropriate corrective actions have been taken;
- To investigate third-party complaints;
- To conduct pre-approval/clearance inspections;
- To inspect new facilities or newly-registered establishments; and
- To inspect facilities under new management and/or ownership.
During an inspection, the FDA collects samples of products and labeling, interviews staff, and inspects facilities, documents, and production processes. The inspectors focus on various “subsystems,” which include: (1) production and process controls, (2) corrective and preventive actions, (3) design controls, (4) management controls, and (5) document controls.
- Production & Process Controls. Production and process controls generally relate to establishing and maintaining procedures to document, monitor, and verify compliance of the manufacture of the device. These apply to all phases of production from initial purchase and receipt of raw materials and/or parts and extending to the sale, distribution, installation and servicing of the finished product.
-
Corrective & Preventive Actions. Corrective and preventive actions generally relate to establishing and maintaining procedures for investigating non-conformances, identifying actions to address these, and verifying that these actions are effective.
-
Design Controls. Design controls generally involve the establishment and maintenance of procedures to control the design of the device to ensure that the specified design requirements are met.
-
Management Controls. Management controls refer to policies, organizational structure, resources, personnel, and internal review of the quality system.
-
Document Controls. Document controls relate to procedures for documenting device and batch/lot-specific specifications and production processes, as well as for document approval, distribution, and changes.
Warning Letters
At the conclusion of an inspection, the FDA provides feedback through written reports known as “Inspectional Observations” or “Form 483.” The reports specify the observed conditions or practices where an FDA-regulated product may be in violation of the FDA’s requirements. This does not constitute a final determination of whether any condition is in violation of the Food Drug and Cosmetic Act (FDCA) or any of its related regulations. Rather, each report serves as a guide for corrective actions. If the FDA does not receive an adequate response, or if a company’s violations are serious enough in nature, the company may receive a Warning Letter from the FDA, have its non- conforming product sequestered, or may be completely shut down.
Warning Letters almost always require a written response, usually within 15 working days. Importantly, Warning Letters indicate violations of regulatory significance that could lead to further enforcement actions if not promptly and adequately corrected. While they are not considered a final regulatory action by the FDA, they serve to establish prior notice. Because Warning Letters are made public and published on the FDA website, they are one of the primary ways in which the FDA seeks to achieve prompt voluntary compliance by the manufacturer. In some exceptional instances, the FDA may bypass the issuance of a Warning Letter and instead take immediate enforcement action. For example, following alleged quality violations at selected manufacturing facilities, Schering Plough agreed to pay a $500 million dollar fine in 2001. Furthermore, its President/Chief Operating Officer resigned, the approval of a pharmaceutical product was delayed for almost a year, and the company’s market value was estimated to have been reduced by more than $30 billion.
Trends in FDA enforcement activities
To identify trends in FDA enforcement activities, the FDA’s databases, the records of FDA’s Center for Devices and Radiological Health (CDRH) inspections, Form 483s and Warning Letters, as well as the specific Quality System Regulations citations in the Warning Letters, were compiled and reviewed. The analysis showed the number of inspections conducted by the CDRH rose sharply from about 2,400 in 2009 to almost 3,400 in 2011 (Figure 1), but has since declined. There is a similar trend for Form 483s and Warning Letters. Furthermore, the number of quality system violations cited in Warning Letters has generally increased; in 2010, 45% of Warning Letters had quality system citations, while from 2011-2014 approximately 75% of Warning Letters included quality system citations. The average number of quality system citations per Warning Letter has also continued to increase from about seven in 2010 to nine in 2014.
-min.jpg?width=488&height=340&name=FDA-warning-letters-1%20(1)-min.jpg)
.jpg?width=485&height=317&name=FDA-warning-letters-2-min%20(1).jpg)
Figure 1. Number of FDA inspections, Form 483s, and Warning Letters between Fiscal Year 2009 and Fiscal Year 2014 (top) and proportion of inspections resulting in the issuance of a Form 483 and Warning Letter (bottom).
Violations related to production and process controls and corrective and preventive actions (CAPAs) account for many citations. In 2014, of Warning Letters with quality system citations, 88% included at least one citation for production and process controls and 86% included at least one citation for corrective and preventive actions. Design controls citations were identified in 59% of Warning Letters.
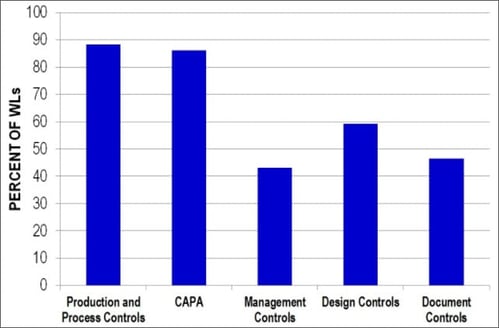
Figure 2. Percent of citations in each quality system subsystem in Fiscal Year 2014. The percent is based on Warning Letters with at least one quality system citation.
Across 2010 through 2014, the most commonly cited regulations for quality system deficiencies in Warning Letters were 21 C.F.R. § 820.100(a) and 198(a) (Table 1). These relate to corrective and preventive actions procedures and complaint filing procedures, respectively. Furthermore, many of the same quality system citations appear as the most frequently cited deficiencies each year, which suggests that the FDA may emphasize specific aspects of the Quality System Regulations over others and that medical device manufacturers should be attentive to these requirements.
Table 1. Top 10 Warning Letter quality system citations
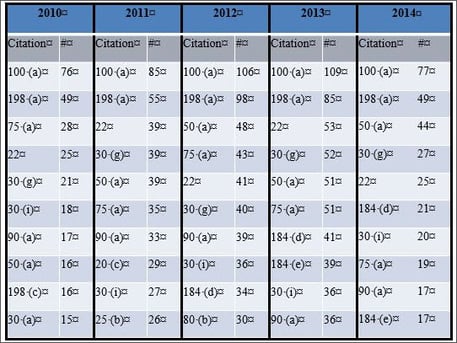
It is noteworthy that two subparts of designs controls (30(g) and 30(i) of 21 C.F.R. § 820.30) are consistently in the top ten quality system citations (Table 1). Section 820.30(g) relates to design validation, which helps ensure that the devices conform to the defined user needs and intended uses, while § 820.30(i) describes the verification, review, and approval of design changes. In 2014, 21 C.F.R. § 820.30(a) and 30(f) were also highly cited in FDA Warning Letters (Figure 3). These describe design procedures (to control the device design to ensure that design requirements are met) and design verification (design output meets design input requirements), respectively. It is likely that design validation is the most frequent design control citation because it may be easily confused with design verification. Design verification refers to whether the product was made right (e.g., were dimensional or strength requirements fulfilled)? On the other hand, design validations refer to whether the right product was made (e.g., if a product was intended to be used laparoscopically, was it evaluated under conditions that simulate a laparoscopic surgery environment)?
The benefits of design controls may include improving the quality of the product, shortening development time by recognizing possible problems earlier and making corrections, lowering manufacturing costs, reducing the risk of product recalls, and minimizing potential products liability exposure.
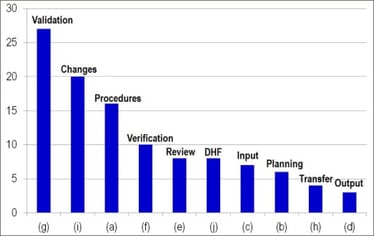
Figure 3. Number of design control (21 C.F.R. § 820.30) citations in each quality system subsystem in Fiscal Year 2014.
Conclusion
Failure to address the FDA’s concerns stated in Warning Letters can lead to enforcement actions that have significant repercussions. The citations in FDA Warning Letters highlight the importance of quality management in the pharmaceutical and medical device industries. Manufacturers are therefore encouraged to take a proactive approach in assessing one’s internal quality management system on an ongoing basis, which can be done by their own staff and/or by an independent third party. These efforts can help reduce the risk of QSR citations by the FDA, as well as the exposure to product recalls and products liability litigation.
Bibliography
- FDA Commissioner’s page (http://www.fda.gov/aboutfda/commiss ionerspage/default.htm)
- U.S. FDA device chief leaves, changes seen (http://www.reuters.com/article/2009/08/11/fda-devices-idUSN1154141520090811)
- Quality system inspection technique (http://www.fda. gov/downloads/ICECI/Inspections/InspectionGuides/UCM085938.pdf) - Guidance for auditing medical device manufacturers against quality system regulation (21 C.F.R. § 820) and related regulations
- FDA inspections, compliance, enforcement, and criminal investigations (http://www.fda.gov/ICECI/Enforcemen tActions/ucm250720.htm)
- Buckley B and Blanks R. Overview of the GxPs for the Regulatory Professional. FDA Regulatory Affairs: A Guide for Prescription Drugs, Medical Devices, and Biologics, 2nd ed. Pisano DJ and Mantus DS (eds.), Informa Healthcare, New York, 2008
- Schering-Plough Enters Into Consent Decree With FDA to Resolve U.S. Manufacturing Issues (http://www.prnewswire.com/news-releases/schering- plough-enters-into-consent-decree-with-fda-to-resolve-us-manufacturing-issues-77491357.html)
Copyright © 2024 - Medmarc